 时刻新闻
时刻新闻
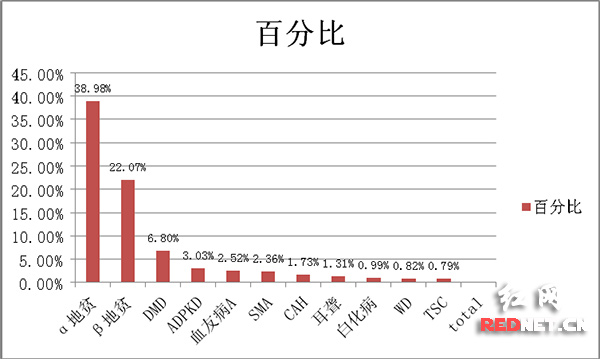
中信湘雅罕见病接诊量前十排行榜
红网时刻2月28日讯(记者 易征洋 通讯员 董雷)2月28日,是国际罕见病日。什么是罕见病?他们究竟有多罕见?是否能够健康生育?这天,作为我国生殖遗传医学领域的翘楚,中信湘雅生殖与遗传专科医院举行系列科普活动,其中公布了医院历年来接诊的罕见病患者及其家系成员中,排名前十位的罕见病名单,地中海贫血、假肥大型进行性肌营养不良症(DMD)、成人多囊肾(ADPKD)占据前三名。
中信湘雅生殖与遗传专科医院院长林戈教授介绍,目前医院已可通过胚胎植入前遗传学诊断/筛查(PGD/PGS)技术帮助罕见病患者实现健康生育,其中包括179种罕见病(189个基因)的PGD技术平台,截至2017年4月,已应用PGD/PGS诞生2380个健康婴儿,在全国处于领先地位。
“罕见病”群体呼吁社会关注
罕见病又称“孤儿病”, 根据世界卫生组织的定义,是指患病率低于0.65‰-1‰的一类疾病,一般为慢性、严重性疾病,多为遗传性,常危及生命。目前,国际确认的罕见病近7000种。中国有大约1000万名罕见病患者,多数罕见病患者对自己的疾病了解不多,国内研究罕见疾病的机构、专家少,且医保目录、现行的医疗保障制度没有涵盖罕见病的治疗和用药,相关的药品研发起步晚,进口药品费用昂贵,部分患者需要终生用药,巨大的治疗费用也给患者家庭带来了沉重的经济负担。例如,罕见病中的重型β-地中海贫血患儿,如果不接受常规输血、铁螯合治疗或造血干细胞移植,通常会在5岁以下死亡,平均寿命约为30岁。
为帮助更多罕见病患者认识疾病,提高防治常识,28日罕见病当天,一批特殊的客人来到中信湘雅生殖与遗传专科医院,向遗传咨询患者义务宣教。他们均是罕见病“资深”病友,他们的到来,一方面希望呼吁社会各界加大对罕见病群体的关注与援助,另一方面也希望能为正在这里治疗的罕见病患者加油鼓气、交流经验。
中信湘雅医院院长林戈介绍,罕见病暂时不能根治,最好的预防办法是防止患儿的出生,可通过产前诊断和植入前遗传学诊断/筛查(PGD/PGS)达到生育健康孩子的目的。为帮助他们实现这一愿望,医院拟启动一项新的援助项目,即向100名PGD患者提供指定助孕药品援助,目前项目适应人群、申报条件、流程等正在制定,近期即将公布。这是医院继为单基因病PGD助孕患者减免2500元费用项目之后,再次推出的又一有力援助举措。
179种罕见病已能实现健康生育
林戈介绍,医院遗传中心一直致力于对遗传性疾病研究,2017年为15000余名患者提供遗传咨询服务,是目前国内最大的细胞和分子遗传学诊断中心之一,也是我国基因诊断数量和种类最多的中心之一。截至目前,中心已建立212种罕见病(单基因病)的基因检测技术,其中179种罕见病(189个基因)已能在医院通过PGD技术实现健康生育。通过孕前基因筛查和PGD技术可有效预防罕见病遗传至下一代,也让已经罹患罕见病的患者有了可以生育健康宝宝的可能。
2017年医院试管婴儿治疗周期超过4.3万例,平均妊娠率达62.4%,医院已累计助孕出生健康婴儿超过12万个,到2017年4月,已应用PGD/PGS技术,诞生了2380个健康婴儿,目前还有1200多例正在妊娠中。帮助众多罕见病家庭实现了健康生育。以假肥大型进行性肌营养不良症(DMD)为例,截至2016年底,已有20个DMD家系在医院通过胚胎植入前遗传学诊断辅助治疗技术生育了健康的孩子。
DMD为X-连锁隐性遗传病,发病率为1/3500,DMD患者起病隐匿,出现明显症状年龄为2~5岁。首发症状为双下肢肌无力,若不治疗干预,DMD患者通常在12岁前失去行走能力,最终因心肺功能衰竭在20~30岁死亡。
武汉的李女士是一名DMD患儿的妈妈。2015年初,她来中信湘雅医院就诊,基因诊断结果提示其DMD致病基因(NM_004006.2)第3-7号外显子杂合缺失,患儿DMD致病基因第3-7号外显子缺失。随后,李女士申请在医院进行DMD-PGD助孕,完成前期的PGD预实验和术前检查后,于2015年6月进入PGD周期,活检4枚囊胚;进行DMD缺失突变检测和连锁分析,2枚为携带者,2枚为正常。正常胚胎继续送检PGS(胚胎植入前遗传学筛查),核型均正常。2016年3月,李女士移植一枚正常胚胎,产前诊断提示正常,2016年12月分娩一健康女婴。
植入前遗传学诊断帮助该家系预防了患儿的出生,避免了孕妇因患病胎儿而行流产的风险,阻断了DMD致病突变在该家系的传递,帮助患者家庭生育了健康的小孩,达到了优生优育的目的。
湖南及周边地区罕见病排行榜出炉
医院遗传中心主任谭跃球教授介绍,为更加直观的展示罕见病科普常识,医院以近年来遗传咨询门诊近28000例罕见病及其家系成员接诊病例数量为基础,组织多名专家整理出了湖南及周边地区常见罕见病排名,并首次向社会公布。
根据医院发布的榜单,地中海贫血、假肥大型进行性肌营养不良症(Duchenne muscular dystrophy, DMD)、成人多囊肾、甲型血友病、脊肌萎缩症、肾上腺皮质增生症、遗传性耳聋、白化病、肝豆状核变性、结节性硬化症占据榜单前十名。这些疾病有哪些特征?是如何遗传的?医院也做出了详细说明。
第一名:地中海贫血
接诊数据:α-地贫10894例、β-地贫6170例
发病特点:地中海贫血遍布全球100多个国家和地区,尤以地中海沿岸国家、中东,北非和东南亚各国多见。中国人群α-地贫、β-地贫、α+β地贫总体发病率分别为是7.88%,2.21%和0.48%。我国南方发病率较高,其中以广东、广西、海南、四川等多见。α-地贫重症患者将在宫内死亡,并危及母亲性命。重型β-地贫孩子如果不接受常规输血、铁螯合治疗或造血干细胞移植,通常会在5岁以下死亡。
致病原因:地中海贫血的特点是由于珠蛋白基因缺陷致使血红蛋白中一种或一种以上珠蛋白链结构异常或合成速率异常造成肽链不平衡,含有这种异常血红蛋白的红细胞变形性降低,寿命缩短,可以提前被人体的肝脾等破坏,导致贫血甚至发育等异常。
典型表现:重型出生数日即出现贫血、肝脾肿大进行性加重,黄疸,并有发育不良,其特殊表现有:头大、眼距增宽、马鞍鼻、前额突出、两颊突出,其典型的表现是臀状头,长骨可骨折。骨骼改变是骨髓造血功能亢进、骨髓腔变宽、皮质变薄所致。少数患者在肋骨及脊椎之间发生胸腔肿块,亦可见胆石症、下肢溃疡。中间型患者轻度至中度贫血,大多可存活至成年。轻型为轻度贫血或无症状,一般在调查家族史时发现。
第二名:假肥大型进行性肌营养不良症(DMD)
接诊数据:1901例
发病特点:假肥大型进行性肌营养不良症(Duchenne muscular dystrophy, DMD, OMIM 310200)和轻型Becker肌营养不良(Becker muscular dystrophy, BMD, OMIM 300376)是最为常见的遗传性肌病,绝大部分是男孩患病,女性患者较少,症状也较男性患者为轻。该病以缓慢进行性加重的对称性肌无力和肌萎缩为特点,可伴有静止性认知障碍和心肌损害,发病率(活产男婴)约为1/3500。
致病原因:DMD/BMD为X-连锁隐性遗传病,致病基因为Dystrophy基因,该基因是目前人类发现的最大的基因,具有突变频率高且突变形式多样的特点,约1/ 3 突变为新生突变。突变类型中基因缺失占55%~65% ,重复占5%~10% ,点突变占25%左右,其他微小缺失和微小重复占8%左右。
典型表现:
1、运动发育较正常儿童晚,如学会走路晚、步态蹒跚、不能跑步、常无故摔倒。在3~5岁时症状逐渐明显,因骨盆带肌力弱,不能跳跃、奔跑,上楼费力,行走姿势异常,腰椎过度前突,骨盆向两侧摆动,呈典型的“鸭步”;病程早期有双下肢腓肠肌假性肥大(脂肪和结缔组织浸润肌肉的结果。);由于腹直肌和髂腰肌无力,患者由仰卧位起立时,先翻身转为俯卧位,然后伸直双臂用双手支撑床面,双腿亦伸直,逐渐用双手扶住膝部,依次向上攀附大腿部,直到立起,这一动作是DMD的特有表现,称为Gower征;萎缩无力肌肉开始主要是大腿和骨盆带肌,逐渐发展至小腿肌、上肢近端、上肢远端肌肉,最后呼吸肌麻痹。腓肠肌肥大常非常显著,其他可出现舌肌、三角肌、臀肌等肌肉肥大。
DMD常伴有心肌损害,累及心室、心房、传导系统。晚期出现心脏扩大、心力衰竭,约10%患者可因心功能不全死亡。此外可出现关节挛缩、足下垂、脊柱侧弯等。多数在12岁左右不能行走,20岁左右因呼吸肌无力、呼吸道感染,引起呼吸肌衰竭死亡。
BMD临床表现与DMD类似,但发病年龄较晚,约为5~15岁,病情较轻,进展速度较慢,12岁以后仍能行走,存活时间较长,部分可接近正常寿命。
第三名:成人多囊肾(ADPKD)
接诊数据:847例
发病特点:常染色体显性遗传性多囊肾疾病(Autosomal dominant polycystic kidney disease,ADPKD),即成人型多囊肾,是以双侧肾脏发生多个囊肿且进行性增大,肾脏结构和功能发生障碍最终导致肾功能衰竭为特征的一组单基因遗传疾病,是人类最常见的常染色体显性遗传病,人群发病率为1/1000。可累及多个系统,如消化系统、心脑血管系统、中枢神经系统、生殖系统、形成肝囊肿、脾囊肿、颅内动脉瘤、心脏瓣膜异常等,此病具有迟发显性,常在成年后发病,但最新研究表明胎儿时期也可发病。
致病原因:已知有PKD1、PKD2、PKD3基因突变可导致ADPKD,85%~90% ADPKD是因PKD1基因突变所致,约10%~15%是由PKD2的突变所致,PKD3基因目前尚未定位。PKD1基因定位于16号染色体,由46个外显子组成,编码产物是由4302个氨基酸残基构成的一种糖蛋白,称为多囊蛋白1(PC1)。PKD2基因定位于4号染色体,由15个外显子组成,编码产物是968个氨基酸残基的多囊蛋白2(PC2)。目前对ADPKD的分子生物学研究证明,多囊蛋白1(PC1)和多囊蛋白2(PC2)在维持细胞内Ca2+的正常浓度中起着重要作用。当PKD1基因或PKD2基因发生突变时,可引起囊泡上皮细胞增殖和囊肿形成等病理改变。
典型表现:常染色体显性遗传性多囊肾病是系统性疾病,多数于30~50岁之间发病,偶可发生于幼年或老年。主要临床表现为腹痛、肾功能损害,若伴发结石或尿路感染,可出现血尿、脓尿、发热、肾区叩击疼痛等症状。1/3患者有肝囊肿,但无肝功能变化。50%~70%患者有高血压,轻到中度的蛋白尿,约有半数患者出现慢性肾功能不全。并发症包括尿毒症、高血压、心脏二尖瓣脱垂、腹壁疝、胰脾囊肿等。
第四名:甲型血友病(血友病A)
接诊数据:704例
发病特点:甲型血友病(hemophilia A,HEMA,OMIM 306700)又称抗血友病球蛋白(antihemophilic globulin, HAG)缺乏症或第VⅢ因子缺乏症。甲型血友病是一种X连锁的隐性遗传病,约占先天性出血性疾病的85%,在男性活婴的发病率约为1/5000,女性主要为携带者,患者少见。
致病原因:编码凝血因子Ⅷ的基因发生突变而导致的该凝血因子功能缺陷或含量不足是该病发生的主要原因。
典型表现:临床表现为自幼反复自发或轻微创伤后全身各部的出血,除皮肤、粘膜出血外,以肌肉、关节出血为特点,反复关节出血往往导致关节畸形,重要脏器出血甚至可危及生命。颅内出血也可发生,也是导致死亡的原因。目前只能靠替代疗法、对症处理等来维持患者的生命,临床上没有找到有效的根治性治疗措施,因此对有生育要求的可疑携带者进行基因诊断,筛查出携带者进行产前诊断,从而降低血友病患儿的出生率是目前针对甲型血友病最行之有效的办法。
第五名:脊肌萎缩症(SMA)
接诊数据:659例
发病特点:脊髓性肌肉萎缩症(Spinal muscular atrophy,SMA)是一类由脊髓前角运动神经元和脑干运动神经核变性导致肌无力、肌萎缩的疾病。根据OMIM收录,脊髓性肌肉萎缩症相关疾病有多种,5q型SMA疾病临床多见,其在活产婴中的发病率为1/10000,携带者频率为1/50。
致病原因:95%的5q型SMA患者是由于SMN1基因第7号外显子纯合缺失导致,约5%的患者是由于SMN1基因点突变所导致。
典型表现:5q型SMA大多数患者为Ⅰ型,症状最为严重。一般在出生6~12个月内发病,表现为出生后哭声微弱,吮奶无力,呼吸与吞咽困难,四肢肌张力极低,常存活至数月即死亡。出生后发病的婴儿在出生时可正常,2-3月后急性发病,表现为肌张力低下、肌萎缩,伴双髋关节屈曲、两腿外展、膝关节屈曲如蛙腿状、腱反射消失,累及延髓者可见舌肌萎缩和纤颤,最后呼吸肌受累,多于病后1-2年内死亡。本病至今尚无特异的有效治疗,主要治疗措施为预防或治疗各种严重肌无力产生的并发症如肺炎、营养不良、骨骼畸形行动障碍和精神社会性问题等。所有SMA患者的肌电图均表现为神经源性损伤。
SMA-Ⅱ型多于1岁内起病,进展缓慢。患儿在6-8个月时生长发育正常多数病例表现以近端为主的严重肌无力,下肢重于上肢;许多Ⅱ型患儿可独坐,少数甚至可以在别人的帮助下站立或行走,但不能独自行走;多发性微小肌阵挛是主要表现;呼吸肌吞咽肌一般不受累,面肌不受累括约肌功能正常。本型具有相对良性的病程。生存期超过4年可存活至青春期后。
SMA-Ⅲ型患者一般起病于儿童期或青少年,一般可存活至成年,肺功能损害是本病常见的死亡原因。临床表现首先是四肢近端肌肉萎缩无力,站立时腹部前突,走路摇摆不定,有Grower征,翼状肩胛,病情发展可影响肢体远端。伴有肌束震颤,部分患者可伴有脊柱侧弯畸形和弓形足等先天畸形。
SMA-IV型, 本病发病平均年龄为30岁,临床表现为缓慢进行性近端肢体萎缩无力,有肌束震颤,可逐渐波及肢体远端,进展缓慢。腱反射减弱或消失,感觉正常,锥体束征阴性,无中枢神经系统功能障碍。
所有SMA患者的肌电图均表现为神经源性损伤。
第六名:先天性肾上腺皮质增生症(CAH)
接诊数据:483例
发病特点:先天性肾上腺增生症(Congenital adrenal hyperplasia, CAH)是一组由于编码皮质激素合成必需酶基因(共7个)突变,导致肾上腺皮质类固醇类激素合成障碍所引起的疾病, 为常染色体隐性遗传。根据缺乏酶的类型和临床症状可分为7型,其中以21-羟化酶缺陷症最为常见,其中以21-羟化酶缺陷症最常见,约占90%以上,其经典型发病率为1.1/10万,非典型发病率可达1/1000。;其次为11β-羟化酶缺陷症,约占5%~8%,其发病率大约1/10万活婴;3β-羟类固醇脱氢酶缺陷症少见,而其他几型罕见。
致病原因:突变导致7个基因编码的酶发生缺陷,导致肾上腺皮质激素合成不足,下丘脑和垂体分别代偿性地增加促肾上腺皮质激素释放激素(CRH)与促肾上腺皮质激素(ACTH)的分泌,从而引起肾上腺皮质增生,以代偿肾上腺皮质激素的合成不足。
典型表现:
1、先天性类固醇21-羟化酶缺陷症
经典型包括极度严重经典型(失盐型)与中度严重型単纯男性化型,非经典型又称轻度型。
(1)失盐型
为临床表现最重的一型,是由于皮质醇与醛固酮缺乏,婴儿早期雄激素分泌过多所致。妊娠期即可发病,出生后表现为皮质醇缺乏症群(低血糖,肾上腺皮质功能降低危象),75%的患者表现出失盐症候群。大部分患者通常在出生后1~4周内出现低钠高钾血症、高肾素血症和低血容量休克等肾上腺危象表现。如果不能得到正确及时的诊治,肾上腺危象会导致患者死亡。患者常于新生儿期出现呕吐、腹泻、代谢性酸中毒等失盐症状,皮肤色素沉着。此外,患者常表现出血醛固酮低,新生儿女性男性化或男性假性性早熟等。对于男性失盐型婴儿问题尤为严重,因为他们没有女性婴儿的外生殖器两性畸形,在这些患儿出现脱水和休克之前医生没有警惕CAH的诊断。随着年龄的增长,在婴幼儿期发生过严重失盐表现的CAH病例钠平衡能力会得以改善,醛固酮合成会更加有效。
(2)单纯男性化型
约占1/4的经典型病例,主要表现为患儿皮质醇缺乏以及雄激素分泌过多。新生儿女性外生殖器男性化,男性阴茎增大和阴囊色素沉着等。与失盐型比较,除没有严重失盐表现外,其他雄激素过多的临床表现大致相同。
(3)非经典型
非经典型较少见,出生后一般无临床症状,随着年龄增大,多在儿童期或成年期,渐渐出现雄激素增高体征,因此也称为迟发型21-羟化酶缺陷症,患者只有轻度雄激素过多的临床表现。女性患者在出生时外生殖器正常或轻度阴蒂肥大,没有外生殖器两性畸形。轻度雄激素过多的症状和体征差异很大,很多受累个体会没有症状。最常见的症状为儿童阴毛提早出现,或年轻女性中表现为严重囊性痤疮、多毛症、多囊卵巢、月经稀发甚至闭经。非经典型21-羟化酶缺陷症女性患者也存在生育能力下降,程度比经典型患者轻。
2、11β-羟化酶缺陷症
高血压是CYP11B缺陷症的特征性表现,与DOC增加引起钠潴留与血容量增加有关。少数患者在婴幼儿期因盐皮质激素缺乏出现高钾血症、低钠血症与低血容量。经典型CYP11B缺陷症患儿亦有女性男性化表現。非经典型患者血压正常或轻度升高,出生时外生殖器一般正常,女性患者在青春期前后出现轻度阴蒂肥大,有些成年妇女可仅有多毛及月经稀发。
第七名:耳聋
接诊数据:366例
发病特点:耳聋是临床上常见的疾病,先天性耳聋(双侧)的发病率为0.1%-0.3%;迟发型耳聋(双侧)美国12-40岁人群的患病率为0.31%-1.6%,40-60岁人群的患病率为6.5%-13.1%。
致病原因:耳聋的致病因素复杂,包括遗传因素、耳毒药物的使用、内耳感染、噪音、衰老、自身免疫反应等。在先天性耳聋中遗传因素占60%左右。遗传性耳聋分为综合性耳聋和非综合性耳聋:前者除耳聋外还伴有眼、骨、肾、或皮肤等器官的病变;后者在临床上只有耳聋症状,占所有遗传性耳聋的70%。根据遗传方式的不同,可以将非综合性耳聋分为常染色体显性(AD)、常染色体隐性(AR)及X-连锁遗传性耳聋(XL),分别占遗传性耳聋的20%、80%、1%和<1%。常染色体隐性遗传性耳聋大部分表现为先天性中度至极重度听力损失,常染色体显性遗传性耳聋大部分表现为迟发型渐进性耳聋,听力损失程度多位中度至重度。
典型表现:常染色体隐性遗传性耳聋大部分表现为先天性中度至极重度听力损失,常染色体显性遗传性耳聋大部分表现为迟发型渐进性耳聋,听力损失程度多位中度至重度。
目前中信湘雅医院针对耳聋进行的基因检测的包括:大前庭导水管扩张综合征相关的SLC26A4基因;GJB2、GJB6、mtDNA(12S rRNA1494/1555位点、tRNA Leu 3243位点和mt-DNA 7445位点)、OTOF(常染色体隐性耳聋9型:)MYO15A、CDH23(AR)、WFS1(遗传性耳聋6/14/38型,呈常染色显性遗传,主要表现为低频感音性神经性耳聋)、MYO6(可导致非综合征型耳聋37型和22型,分别呈AR和AD);KCNQ4基因突变导致非综合性耳聋2A型,呈AD,表现为渐进性感音神经性听力损失,一般5-15岁发病; MYH14基因突变导致非综合性耳聋4A型,呈AD,表现为渐进性感音神经性听力损失,一般20岁左右发病;PCDH15基因可导致非综合征耳聋23型和Usher综合征1F型,均呈常染色体隐性遗传, DFNA5导致常染色体显性遗传性耳聋5型。
第八名:白化病
接诊数据:278例
发病特点:白化病(albinism)是一组黑色素生成障碍性遗传病,在世界范围内白化病的发病率约为1/17000,在中国其发病率约为 1/20000~1/10000。该病具有高度遗传异质性,。
致病原因:目前已明确的引起人类白化病的基因有几十个,大致分为以下两大类:非综合型(Nonsyndromic)和综合型(Syndromic)。
非综合型(Nonsyndromic):仅有眼、皮肤和毛发受累的典型症状。
(1)眼皮肤白化病:呈常染色体隐性遗传,如OCA1,OCA2,OCA3,OCA4。
(2)眼白化病:呈X连锁隐性遗传,如眼白化病(OA1)致病基因GPR143。
2、综合型(Syndromic):具有部分白化病典型临床特征外,还伴有其他器官、组织的病变,如具有出血倾向的综合征。
(1)Hermansky-Pudlak综合征(HPS):常染色体隐性遗传,如HPS1~HPS7。
(2)Chediak-Higashi综合征(CHS):常染色体隐性遗传,如CHS1。
(3)Griscelli综合征(GS):常染色体隐性遗传,如GS1,GS2,GS3。
(4)Usher综合征(USH):常染色体隐性遗传,如USH1B。
典型表现:皮肤、毛发和(或)眼部的色素缺乏,伴随有畏光、斜视、眼球震颤和视力低下等眼部症状。
第九名:肝豆状核变性(Wilson病,WD)
接诊数据:230例
发病特点:是一种主要累及肝脏和神经系统的铜代谢紊乱性疾病,是由于WD蛋白的功能缺陷所致的一种常染色体隐性遗传病。发病率1/50000~1/100000,致病基因携带率约为1/90。
致病原因:由ATP7B基因突变导致机体铜代谢异常,过量的铜沉积在肝脏和脑等组织中所引起的一系列临床表现的综合征;ATP7B基因定位于13q14.3,含有21个外显子和20个内含子,近年来有研究发现除ATP7B以外其他基因如COMMD1、XIAP、Atox1等也与该病相关。
典型表现:进行性加重的锥体外系症状、角膜色素环、肝硬化、精神症状及肾功能损害等。根据中华医学会神经病学分会帕金森病及运动障碍学组《肝豆状核变性的诊断与治疗指南》,临床分型如下:
1、肝豆状核变性肝型:①持续性血清转氨酶增高;②急性或慢性肝炎;③肝硬化(代偿或失代偿);④暴发性肝功能衰竭(伴或不伴溶血性贫血)。
2、肝豆状核变性脑型:①帕金森综合征;②运动障碍:扭转痉挛、手足徐动、舞蹈症状、步态异常、共济失调等;③口-下颌肌张力障碍:流涎、讲话困难、声音低沉、吞咽障碍等;④精神症状。
3、肝豆状核变性其他类型:以肾损害、骨关节肌肉损害或溶血性贫血为主。
4、肝豆状核变性混合型:以上各型的组合。
第十名:结节性硬化症
接诊数据:220例
发病特点:结节性硬化症(TSC)又称Bourneville病,是一种常染色体显性遗传的神经皮肤综合征,也有散发病例,多由外胚叶组织的器官发育异常,可出现脑、皮肤、周围神经、肾等多器官受累。病率约为1/6000活婴,男女之比为2:1。儿童期发病,男多于女。结节性硬化症(TSC)属于慢性罕见病,是一种累及多系统的常染色体显性遗传性疾病,长期以来缺乏根治方法。全球约有100万TSC患者,在中国估计目前有14.25万~20.00万例。
致病原因:该病为遗传病,根据基因定位可分为四型:TSC1、TSC2、TSC3、TSC4。TSC1和TSC2突变分别引起错构瘤蛋白和结节蛋白功能异常,影响其细胞分化调节功能,从而导致外胚层、中胚层和内胚层细胞生长和分化的异常。
典型表现:结节性硬化症的临床表现多样,癫痫、智力低下、皮脂腺腺瘤是典型的“三联征”。一半以上的患者会出现Pringle皮脂腺腺瘤,病理显示为一种血管纤维瘤,大多在3-10岁出现,多分布在颜面部,随年龄增长而逐渐增大为结节。40-60%的患者会出现不同程度的智力低下,90%的患者可能会出现癫痫。此外,还可能会出现甲周纤维瘤、鲨革样斑、条形叶状白班、肾脏病变、室管膜下结节、单发或多发的心脏横纹肌瘤、肺淋巴管瘤肌瘤病、眼视网膜病等。
来源:红网
作者:易征洋
编辑:曼曼
本文为红网原创文章,转载请附上原文出处链接和本声明。
本文链接:https://health.rednet.cn/content/2018/02/28/417730.html